Import Files
Import Sequence Files
Krait2 supports fasta and fastq formatted sequence files as well as the gzip compressed fasta/q files.
Go to File menu, click Import sequence files… to open file select dialog, select sequence files, and then click Open to import files.
Or, you can go to File menu, click Import sequence files from folder… to open folder select dialog, select a folder, and then click Select Folder to import all sequence files in that folder.
Import Annotation Files
Currently, Krait2 supports GTF and GFF formatted annotation files as well as the gzip compressed gtf/gff files.
Go to File menu, click Import annotation files… to open file select dialog, select annotation files, and then click Open to import files.
Or, you can go to File menu, click Import annotation files from folder… to open folder select dialog, select a folder, and then click Select Folder to import all annotation files in that folder.
Note
The annotation file name must be consistent with the corresponding sequence file. When the annotation file is imported, the krait2 will automatically match it with the sequence file. If a match is found, the sequence file name in the input file list will be bolded.
For example, example.fa.gz will match with example.gtf.gz, keep the names before the first dot are the same, they can be recognized.
Search for Repeats
Prior to search for repeats, you can go to Edit menu, click Repeat search settings to open search parameter setting dialog to set corresponding paramters.
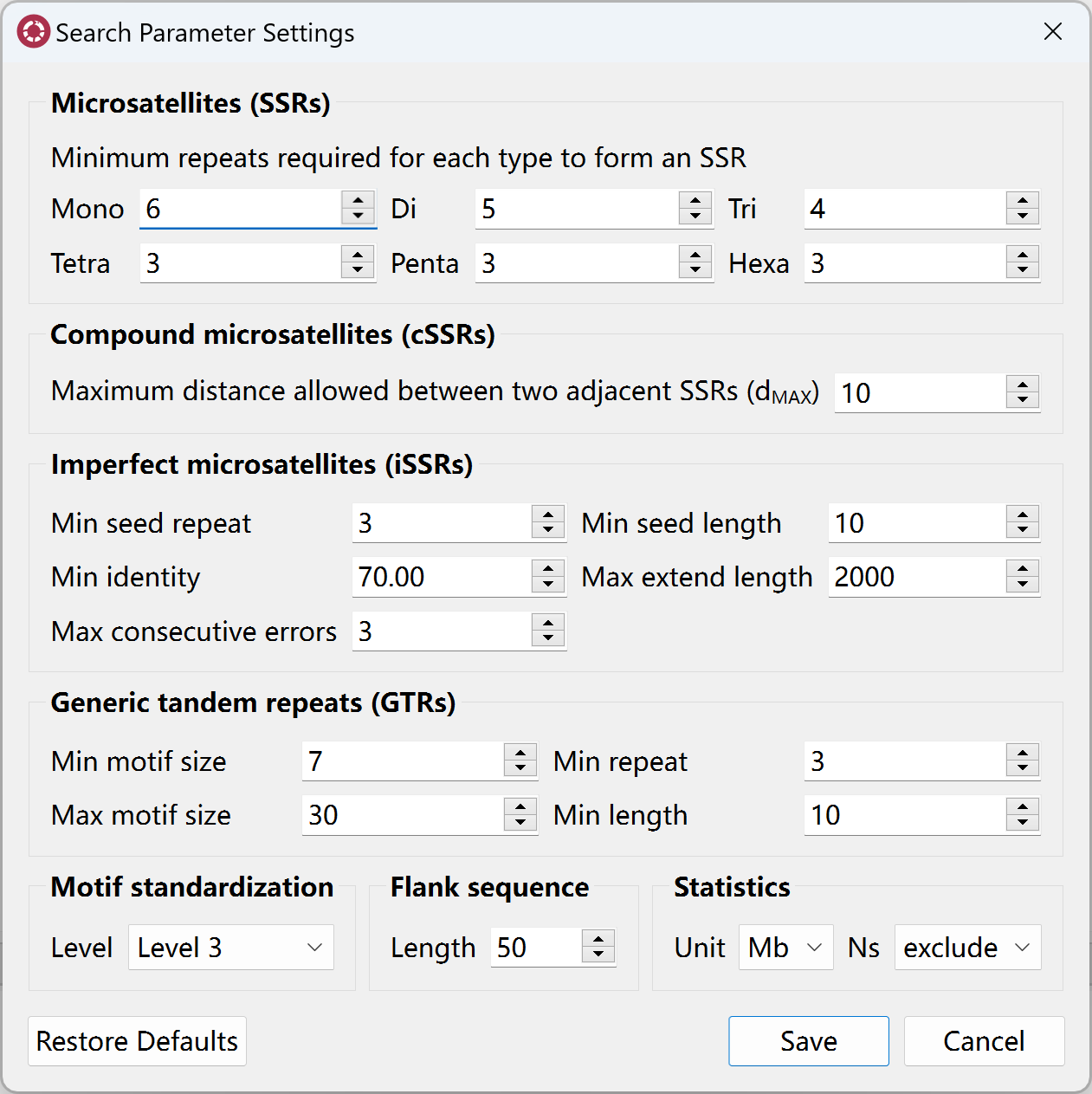
Search for Perfect SSRs
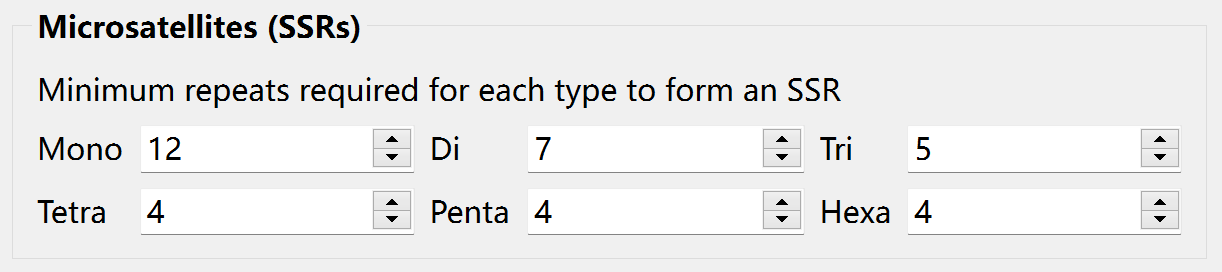
Before searching for perfect SSRs, you can set the minimum tandem repeat number for mono-, di-, tri-, tetra-, penta-, hexa-nucleotide SSRs, separately.
Go to Toolbar, and then click  to start search for perfect SSRs. After searching, you can click the file in
to start search for perfect SSRs. After searching, you can click the file in input file list to view results in SSRs table.
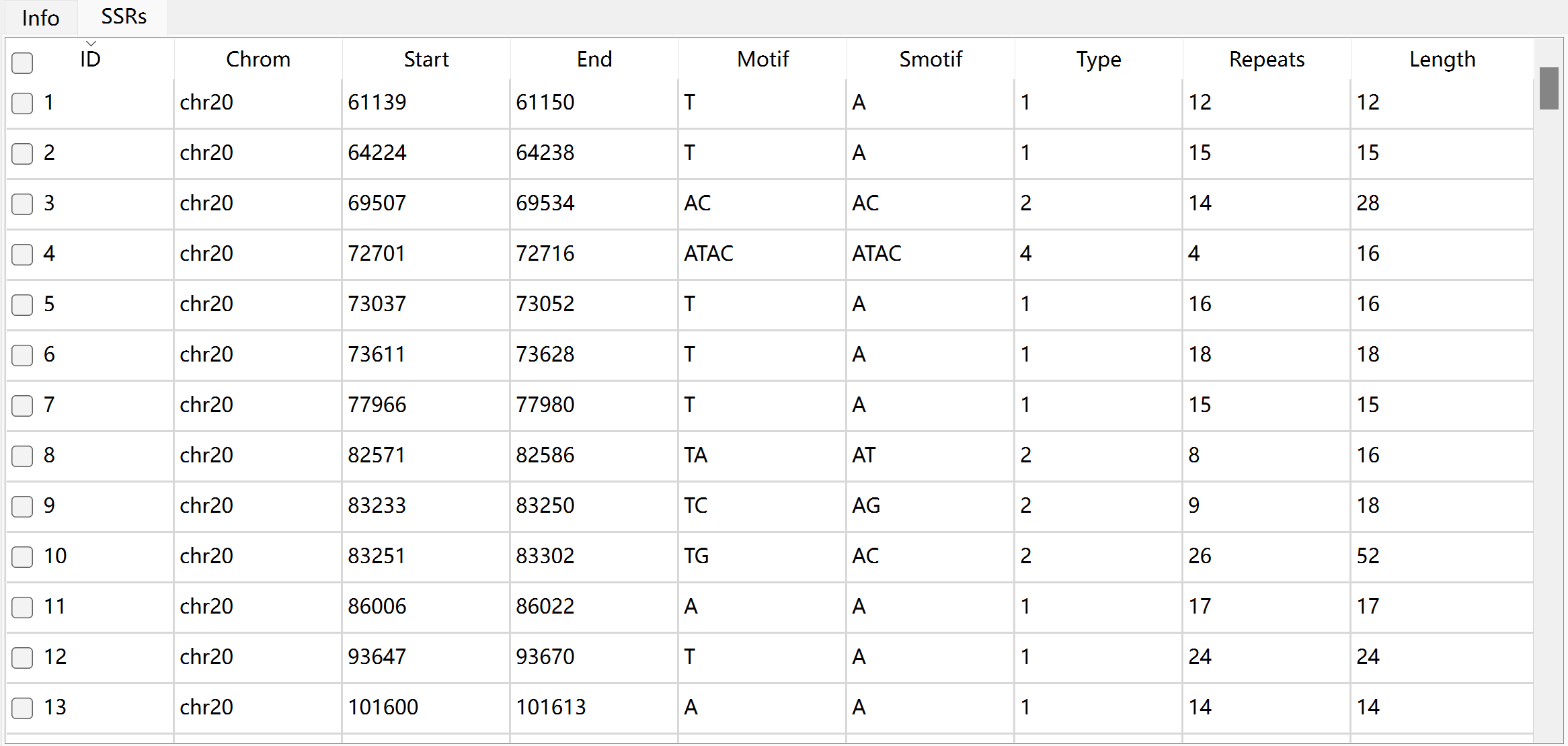
The description of each column in SSRs table:
Column |
Description |
|---|---|
ID |
unique identifier generated by Krait |
chrom |
the name of sequence where SSR was found |
start |
start position of SSR in original sequence, 1-based |
end |
end position of SSR in original sequence, 1-based |
motif |
repeat unit of SSR |
smotif |
the standardized motif |
type |
SSR type or motif length |
repeats |
number of repeats |
length |
the length of SSR (bp) |
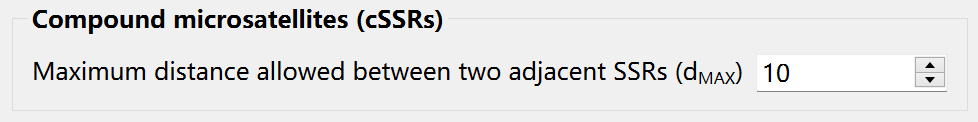
Search for Compound SSRs
Compound SSRs (cSSRs) refer to regions of DNA where two or more different types of SSRs are adjacent to each other. Before searching, you can set the maximum distance allowed between two adjacent SSRs:
Go to Toolbar, and then click  to start search for compound SSRs. After searching, you can click the file in
to start search for compound SSRs. After searching, you can click the file in input file list to view results in cSSRs table.
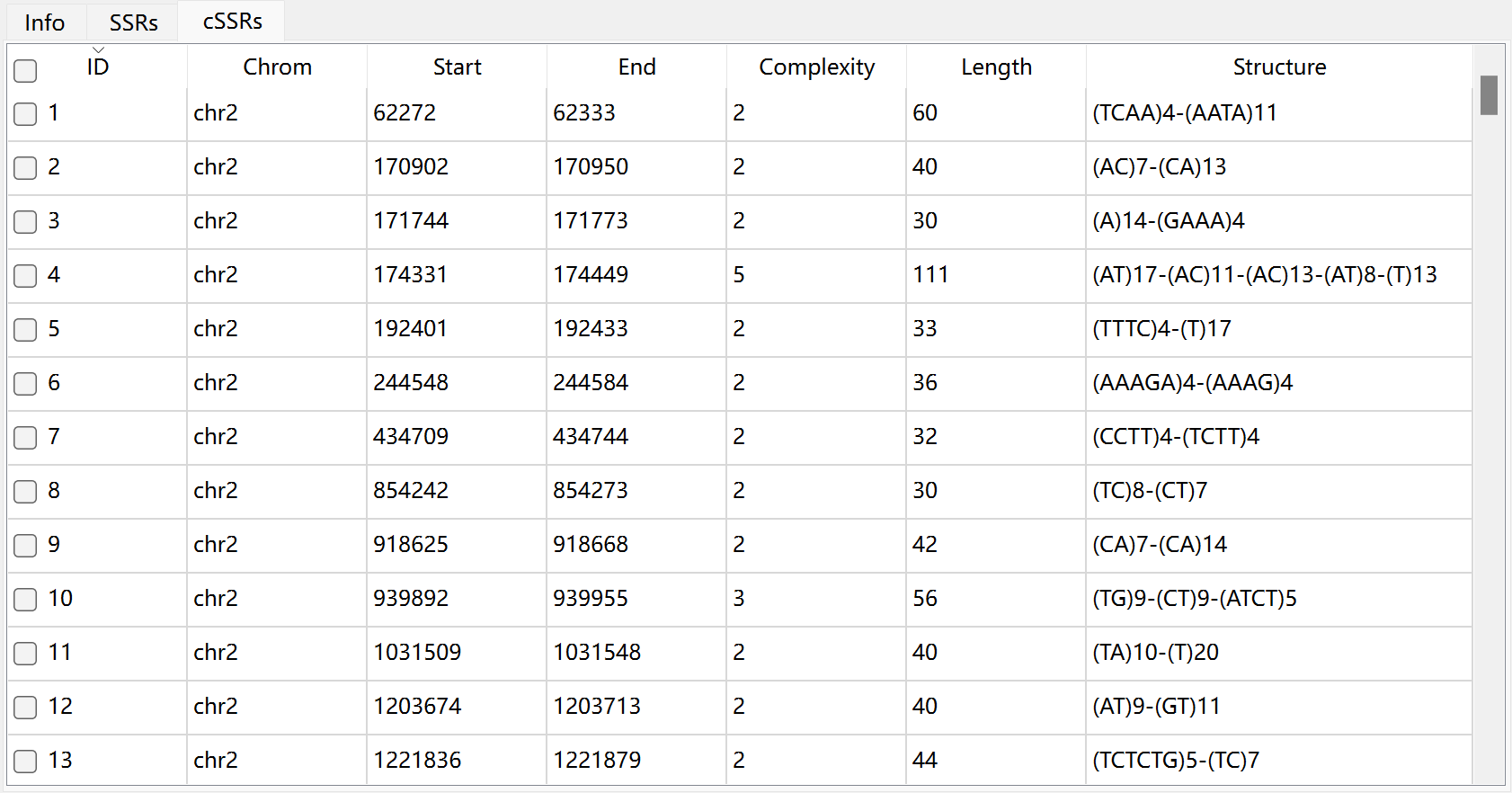
The description of each column in cSSRs table:
Column |
Description |
|---|---|
ID |
unique identifier generated by Krait |
chrom |
the name of sequence where cSSR was found |
start |
start position of cSSR in original sequence, 1-based |
end |
end position of cSSR in original sequence, 1-based |
complexity |
the number of individual SSR in a cSSR |
length |
the length of cSSR (bp) |
structure |
the components of cSSR |
Search for Imperfect SSRs
Imperfect SSRs (iSSRs) are microsatellites where the repetitive DNA sequence is interrupted by one or more non-repetitive nucleotides. The iSSRs allow substitution, insertion and deletion in the sequence. Before searching, you can set the corresponding parameters:
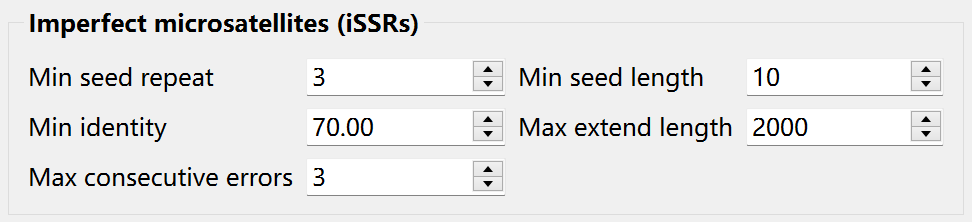
Krait2 will firstly find a seed which is like a perfect SSR meeting the minimum seed repeat and minimum seed length. And then the seed will be extended to left and right flank by aligning flanking sequences to its perfect counterpart. When the extending length exceeds the predefined maximum extend length or the consecutive alignment error exceeds the predefined maximum consecutive errors, the extending will be stopped.
Go to Toolbar, and then click  to start search for imperfect SSRs. After searching, you can click the file in
to start search for imperfect SSRs. After searching, you can click the file in input file list to view results in iSSRs table.
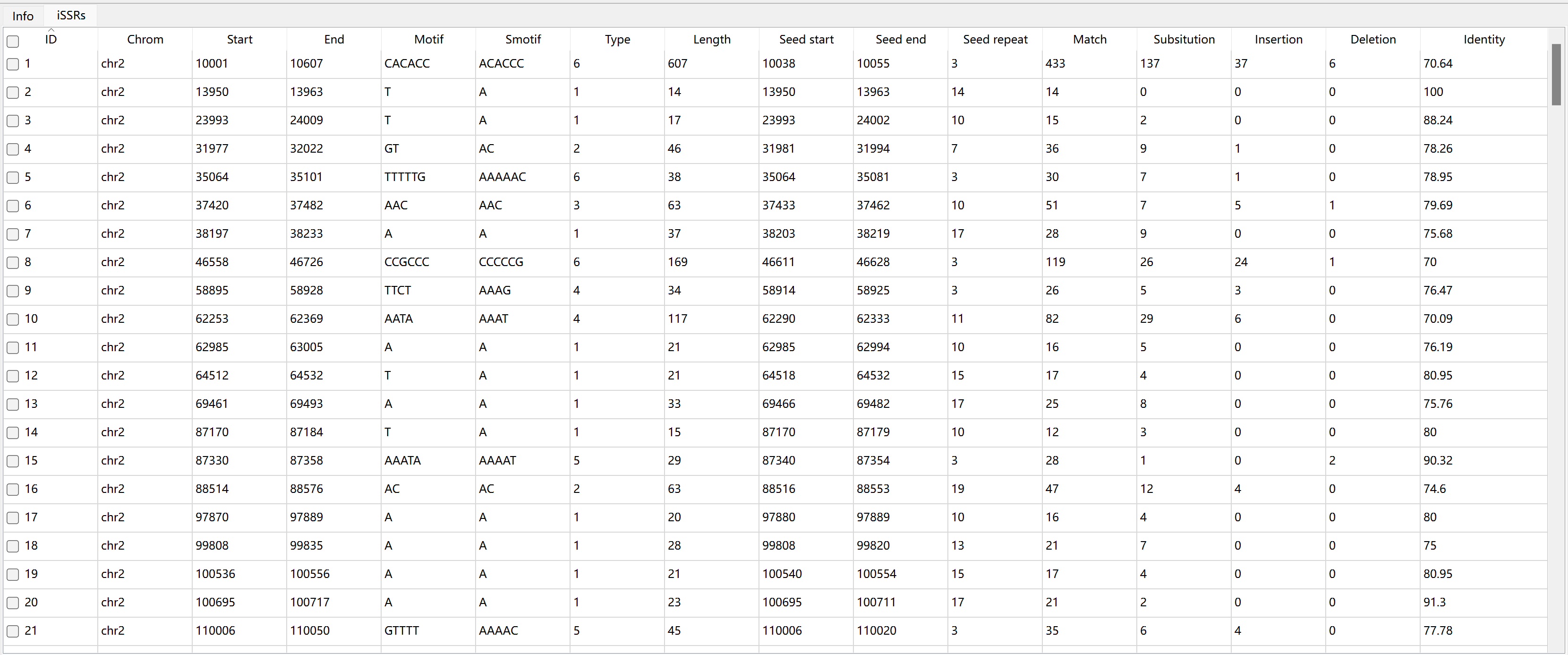
The description of each column in iSSRs table:
Column |
Description |
|---|---|
ID |
unique identifier generated by Krait |
chrom |
the name of sequence where SSR was found |
start |
start position of iSSR in original sequence, 1-based |
end |
end position of iSSR in original sequence, 1-based |
motif |
repeat unit of iSSR |
smotif |
the standardized motif |
type |
iSSR type or motif length |
length |
the length of iSSR (bp) |
seed start |
seed start position in sequence |
seed end |
seed end position in sequence |
seed repeat |
seed repeat number |
match |
number of matches |
substitution |
number of substitutions |
insertion |
number of insertions |
deletion |
number of deletions |
identity |
the alignment identity |
Search for Generic Tandem Repeats
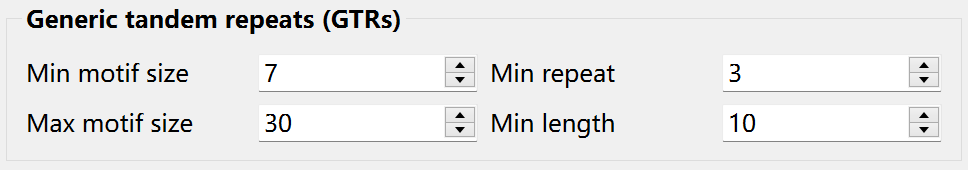
Krait2 allows users to search for generic tandem repeats (GTRs) with motif size <= 1000 bp. Before searching, you can set the minimum motif size, maximum motif size, minimum repeats and minimum length:
Go to Toolbar, and then click  to start search for GTRs. After searching, you can click the file in
to start search for GTRs. After searching, you can click the file in input file list to view results in GTRs table.
The description of each column in GTRs table:
Column |
Description |
|---|---|
ID |
unique identifier generated by Krait |
chrom |
the name of sequence where GTR was found |
start |
start position of GTR in original sequence, 1-based |
end |
end position of GTR in original sequence, 1-based |
type |
GTR type or motif length |
repeats |
number of repeats |
length |
the length of GTR (bp) |
motif |
repeat unit of GTR |